

ด้วยกระแสความนิยมของการเลือกสูบบุหรี่ไฟฟ้าในปัจจุบันทำให้อุปกรณ์และวัสดุที่ผลิตบุหรี่ไฟฟ้าได้รับความนิยมมากในปัจจุบันและหากคุณเป็นหนึ่งในคนที่สนใจจะเลิกสูบบุหรี่ไฟฟ้าแต่ยังตัดสินใจไม่ได้ว่าจะเลือกสูบบุหรี่ประเภทไหนก่อนที่คุณจะเข้าไปเลือกซื้อบุหรี่ไฟฟ้าจำเป็นอย่างยิ่งที่คุณจะต้องเข้าไปทำความรู้จักกับแต่ละรูปแบบของบุหรี่ไฟฟ้าให้ดีเพื่อที่จะช่วยทำให้คุณตัดสินใจได้ง่ายมากขึ้นว่าคุณเหมาะที่จะสูบบุหรี่ไฟฟ้ารูปแบบใดประเภทใดรวมถึงมีน้ำยาบุหรี่ไฟฟ้าแบบไหนให้ใช้งาน ซึ่งแน่นอนว่าในปัจจุบันแม้บุหรี่ไฟฟ้าจะเป็นบุหรี่ที่ได้รับความนิยมมากที่สุดแต่บางครั้งอุปกรณ์ที่ใช้ในการสูบไม่ว่าจะเป็นน้ำยาบุหรี่ไฟฟ้าหรืออุปกรณ์อื่นๆล้วนแต่มีเป็นของรุ่นบุหรี่ไฟฟ้าเองดังนั้นก่อนที่จะตัดสินใจเลือกซื้อตัวบุหรี่ไฟฟ้าประเภทใดก็ตามควรที่จะทำความรู้จักเกี่ยวกับวัสดุอุปกรณ์ที่ใช้เพื่อลดปัญหาต้นทุนที่สูงมากเกินไปทำให้ไม่คุ้มค่ากับค่าใช้จ่ายหรือรายได้ที่คุณได้รับ เอาเป็นว่าถ้าใครสนใจต้องการเลือกซื้อน้ำยาบุหรี่ไฟฟ้าหรืออุปกรณ์บุหรี่ไฟฟ้าในไลน์สามารถเข้ามาดูรายละเอียดเพิ่มเติมได้ที่หน้าเว็บ podbkk podbkk เว็บสำหรับขายน้ำยาบุหรี่ไฟฟ้า ภายในเว็บ podbkk เรามีหลากหลายน้ำยาบุหรี่ไฟฟ้าให้คุณได้ลองเข้ามาทำความรู้จักและใช้งานมีจำนวนนิโคตินที่คุณสามารถเติมได้แตกต่างกันออกไปขึ้นอยู่กับความสนใจและความต้องการของลูกค้าเองว่าอยากได้จำนวนไหนรวมถึงราคาของน้ำยาบุหรี่ไฟฟ้าในปัจจุบันก็มีหลากหลายเรทราคาขึ้นอยู่กับความต้องการของลูกค้าเองว่าจะใช้บริการและเลือกซื้อน้ำยาบุหรี่ไฟฟ้าประเภทไหนให้กับตัวบุหรี่ของคุณ podbkk เราเป็นเว็บสำหรับขายบุหรี่ไฟฟ้ามีหลากหลายอุปกรณ์โดยเฉพาะน้ำยาบุหรี่ไฟฟ้าที่คุณสามารถเข้ามาเลือกซื้อและทำความรู้จักด้วยได้พร้อมรับโปรโมชั่นพิเศษที่จะช่วยทำให้คุณประหยัดเงินในการเลือกซื้อน้ำยาบุหรี่ไฟฟ้าได้มากขึ้นหากคุณต้องการทราบรายละเอียดเพิ่มเติมก่อนที่จะเลือกซื้ออุปกรณ์เหล่านี้สามารถเข้าไปทำความรู้จักได้ที่หน้าเว็บpodbkk เรามีทีมงานมืออาชีพที่พร้อมให้คำแนะนำและให้ข้อมูลเกี่ยวกับรูปแบบน้ำยาบุหรี่ไฟฟ้าและตัวบุหรี่ไฟฟ้าแต่ละประเภทเพื่อที่จะช่วยทำให้คุณตัดสินใจเลือกซื้ออุปกรณ์เหล่านี้ได้ง่ายมากขึ้น หลายคนเชื่อว่าการสูบบุหรี่ไฟฟ้าจะทำให้คุณสามารถลดหรือเลิกบุหรี่ซองได้มีหลายคนที่ประสบความสำเร็จจากการเข้ามาสูบบุหรี่ไฟฟ้าแต่ก็มีอีกเป็นจำนวนมากที่ไม่ประสบความสำเร็จแถมยังทำให้คุณติดบุหรี่ไฟฟ้าเพิ่มมากขึ้นดังนั้นก่อนที่คุณจะเข้ามาใช้บริการใดๆหรือมั่นใจว่าจะเข้ามาสูบบุหรี่ไฟฟ้า ควรลองเช็คความต้องการของตัวเองให้ดีว่าทำไมคุณถึงควรสูบบุหรี่ไฟฟ้าคุณพร้อมที่จะรับในค่าใช้จ่ายต่างๆที่มาพร้อมกับการซื้อเสื้อสูบบุหรี่ไฟฟ้าได้หรือไม่ เพราะการสูบบุหรี่ไฟฟ้านอกจากจะทำให้คุณต้องแบ่งเงินสำหรับการเลือกซื้ออุปกรณ์แล้วคุณยังต้องแบ่งเงินสำหรับการจ่ายค่าอาหารต่างๆอีกด้วย ชื่อหมายความว่าคุณจะต้องแบกรับค่าใช้จ่ายสำหรับการสูบบุหรี่ไฟฟ้าเพิ่มมากขึ้นสำหรับใครที่กังวลเกี่ยวกับปัญหาทางการเงินการตัดสินใจจะเลือกสูบบุหรี่ไฟฟ้าและพร้อมเลือกซื้ออุปกรณ์ที่ใช้ในการสูบบุหรี่ไฟฟ้าอาจจะทำให้คุณมีปัญหาทางการเงินตามมาได้ดังนั้นก่อนที่คุณจะเข้ามาเลือกซื้อน้ำยาบุหรี่ไฟฟ้าหรืออุปกรณ์ใดๆตรวจเช็คความต้องการแท้จริงของคุณให้เจอ

เกษตร ทำไมควรเลือกใช้ “เครื่อง คัด ข้าวเปลือก”เกษตร ทำไมควรเลือกใช้ “เครื่อง คัด ข้าวเปลือก”
เกษตรนั้นถือว่าเป็นอีกหนึ่งในอาชีพหลัก ๆ ของคนไทยเลยก็ว่าได้ เพราะว่าคนไทยนั้นประกอบอาชีพเกษตรเกือบจะ 60 % ของประเทศ ซึ่งอุตสาหกรรมข้าวเปลือกเองก็เป็นอุตสาหกรรมขนาดใหญ่อย่างมากด้วยเช่นกันนั้นจึงทำให้ ปัจจุบันนั้นมีเครื่องจักรที่หลากหลาย ให้เกษตรนั้นได้มีเครื่องที่ใช้ประโยชน์ได้มากขึ้นอย่างหลากหลายมากยิ่งขึ้น ซึ่งหนึ่งในเครื่องจักร ที่มีการใช้งานอย่างหลากหลายอย่างมากนั้นคือ เครื่อง คัด ข้าวเปลือก เป็นอีกหนึ่งในเครื่องมืออุตสาหกรรม ที่มีการใช้งานที่หลากหลายอย่างมากด้วยเช่นกัน ดังนั้นในบทความนี้เราจะมาดูกันดีกว่านะครับว่า เครื่อง คัด ข้าวเปลือก นั้นจะมีความสำคัญอะไรบ้างกับเกษตร ช่วยการแยกสิ่งสกปรกออกจากข้าว สิ่งสกปรกนั้นเป็นสิ่งที่จะติดมากับข้าวสาร และ ข้าวเปลือกก่อนที่จะส่งออก ซึ่งนั้นอาจจะทำให้ราคาของข้าวนั้นอาจจะลดลงได้อย่างมากนะครับหากว่าปล่อยมีให้มีสิ่งสกปรก ดังนั้นหนึ่งในข้อดีอย่างมาก เครื่อง
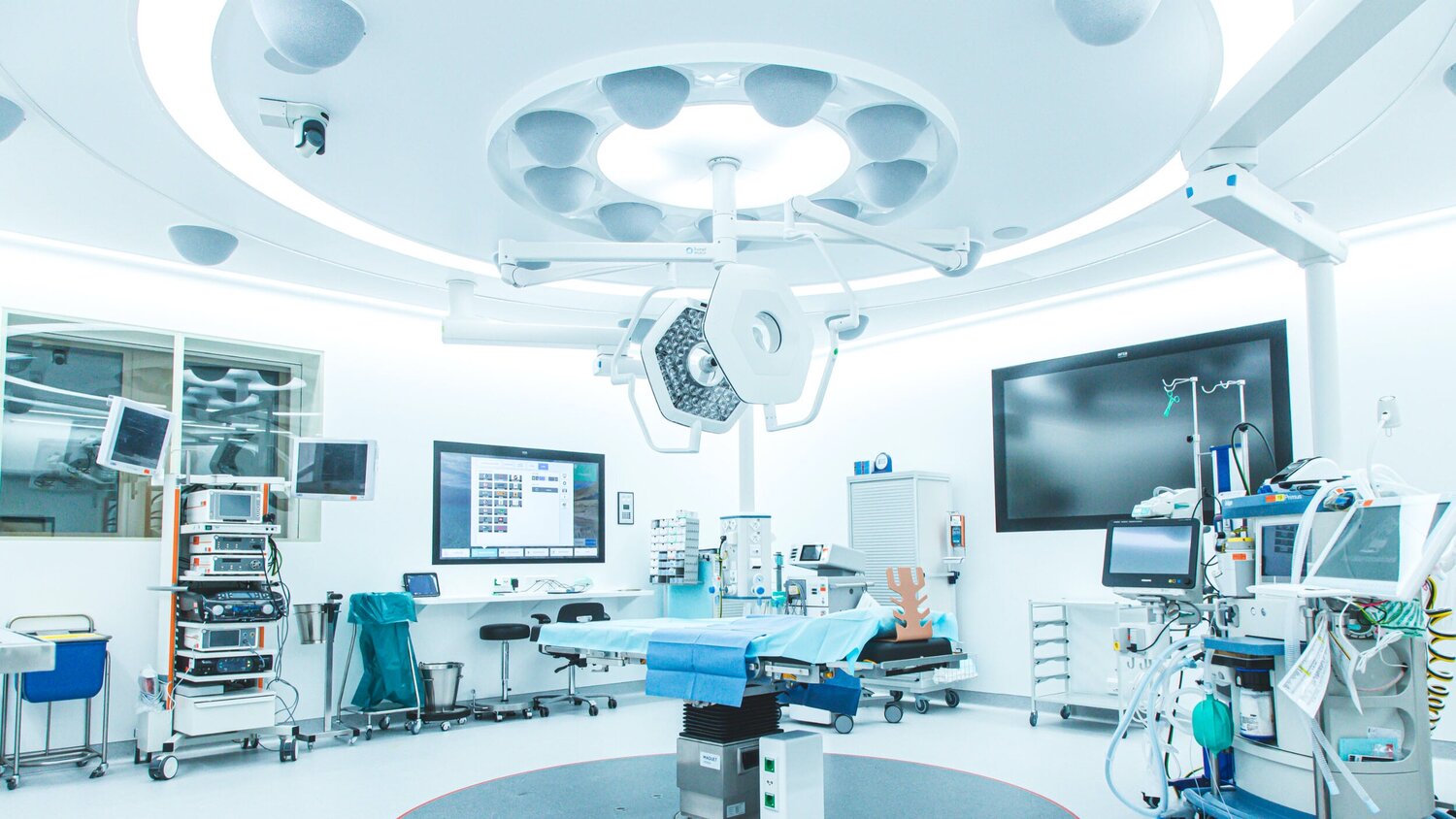
สถานที่ไหนบ้าง “ถ้าหากว่าไม่มีการควบคุมความชื้น” จะเป็นอันตรายอย่างมากสถานที่ไหนบ้าง “ถ้าหากว่าไม่มีการควบคุมความชื้น” จะเป็นอันตรายอย่างมาก
ปัจจุบันนั้นเรื่องของความชื้นนั้นเป็นเรื่องที่สำคัญอย่างมาก และ หลายคนนั้นอาจจะไม่เคยได้รับรู้มาก่อนถึงความสำคัญของ ความชื้น ซึ่งในพื้นที่บางพื้นที่นั้นจะต้องมีการติดตั้ง เครื่อง ควบคุม อุณหภูมิ และ ความชื้น ไว้ด้วยเช่นกัน เพื่อป้องกันในเรื่องของความอันตรายของชีวิต ดังนั้นในบทความนี้เราจะมาดูกันดีกว่านะครับว่าในพื้นที่ไหนบ้างที่มีการควบคุมความชื้นไว้อย่างเคร่งครัด ไม่อย่างนั้นอาจจะเป็นอันตรายต่อชีวิตได้ด้วยเช่นกัน มาลองดูกันดีกว่านะครับว่าจะมีพื้นที่ไหนบ้าง ห้องผ่าตัด หลายคนนั้นอาจจะเคยเห็นในห้องผ่าตัดเพียงผิวเผิน แต่ว่าในความจริงแล้วห้องผ่าตัดนั้นจะต้องมีการออกแบบด้วยวิศกร และ ที่สำคัญนั้นจะต้องมีการออกแบบโดยผู้เชี่ยวชาญด้วยเช่นกันนั้นก็เพราะว่าในห้องผ่าตัดนั้นถ้าหากว่าไม่มีการออกแบบที่ดีนั้นอาจจะทำให้มีความชื้นสัมพันธ์มากเกินไป และ กลั่นตัวเป็นไอน้ำได้ด้วยเช่นกัน ซึ่งจะเป็นอันตรายอย่างมากที่จะต้องระมัดระวังไว้ ดังนั้นภายในห้องผ่าตัดนั้นจึงมีความสำคัญอย่างมาก ที่จะต้องระมัดระวังไว้เพื่อไม่ให้เกิดอันตรายต่อชีวิตของผู้ป่วย ซึ่งเป็นเรื่องที่สำคัญอย่างมากที่เรานั้นอาจจะต้องรู้ไว้เลยก็ได้นะครับ ห้องรักษาผู้ป่วย โรคตา โรคทางเดินหายใจ

เช็คเบี้ยประกัน ก่อนเลือกซื้อความคุ้มครองจากประกันรถยนต์เช็คเบี้ยประกัน ก่อนเลือกซื้อความคุ้มครองจากประกันรถยนต์
เพราะการซื้อความคุ้มครองจากประกันรถยนต์ในปัจจุบันถือว่าเป็นเรื่องง่ายและสะดวกอย่างมากเชื่อว่าหลายๆคนให้ความสนใจและเลือกเข้ามาเลือกซื้อประกันรถยนต์การเพิ่มมากขึ้นแน่นอนว่าก่อนที่คุณจะเข้ามาเลือกซื้อประกันรถยนต์การทำความรู้จักกับแต่ละรูปแบบประกันรถยนต์ถือว่าเป็นเรื่องสำคัญอย่างยิ่งโดยเฉพาะการเช็คเบี้ยประกันว่าผู้เอาประกันนั้นต้องการรูปแบบประกันแบบไหนให้ความคุ้มครองอย่างไรตอบโจทย์ความต้องการหรือไม่รวมถึงราคาเบี้ยประกันที่คุณกำลังมองหาอยู่นั้นตอบโจทย์ความสามารถในการจ่ายประกันของผู้เอาประกันด้วยหรือไม่โดยทั้งนี้หากคุณต้องการข้อมูลเพื่อเช็คเบี้ยประกันสามารถเข้ามาสอบถามได้ที่เว็บรู้ใจของเราเรามีหลากหลายบริการประกันรถยนต์ประกันมอเตอร์ไซค์ให้คุณได้เลือกซื้อรวมถึงบริการเช็คเบี้ยประกันที่จะช่วยทำให้คุณเข้าถึงรูปแบบความคุ้มครองจากประกันที่คุณต้องการได้ง่ายมากขึ้นเอาเป็นว่าถ้าใครพร้อมแล้วก็ลองเข้ามาทำความรู้จักและเลือกซื้อรวมถึงใช้บริการเช็คเบี้ยประกันที่เว็บรู้ใจกันได้เลย บริการเช็คเบี้ยประกันที่เว็บรู้ใจ เพราะเว็บรู้ใจของเราเป็นเว็บที่เปิดให้บริการมานานมีหลากหลายรูปแบบความคุ้มครองจากประกันรถยนต์ประกันรถมอเตอร์ไซค์ประกันชีวิตประกันสุขภาพและรูปแบบความคุ้มครองอื่นๆที่จะช่วยทำให้ผู้เอาประกันได้รับการดูแลมีเกราะป้องกันในการใช้ชีวิตเพิ่มมากขึ้นแต่แน่นอนว่าบริการเช็คเบี้ยประกันนี้คุณสามารถเข้ามาหาข้อมูลเพิ่มเติมได้ที่หน้าเว็บรู้ใจของเราตลอด 24 ชั่วโมงเพราะหน้าเว็บของเรามีหลากหลายข้อมูลเกี่ยวกับรูปแบบประกันให้คุณได้เข้ามาทำความรู้จักและอ่านรายละเอียดเพื่อที่จะทำให้คุณเข้าใจในรูปแบบความคุ้มครองที่คุณกำลังมองหาอยู่ได้ง่ายมากขึ้นด้วยข้อมูลต่างๆเหล่านี้คุณไม่ต้องเสียเวลาเข้ามาสอบถามกับทีมงานขอเว็บรู้ใจของเราเพราะคุณสามารถเข้าไปทำความรู้จักเองได้ที่หน้าเว็บว่าจะเป็นรูปแบบความคุ้มครองข้อยกเว้นความคุ้มครองเงื่อนไขกรมธรรม์ประกันที่คุณสนใจตลอดจนบริการเช็คเบี้ยประกันต่างๆเหล่านี้คุณก็สามารถทำได้เพียงเข้ามาที่เว็บรู้ใจ นอกเหนือจากนี้แล้วหากคุณต้องการเลือกซื้อรูปแบบการใดๆก็ตามสามารถเข้ามาใช้บริการเปรียบเทียบราคาเบี้ยประกันที่จะช่วยทำให้คุณประหยัดเงินในการเลือกซื้อความคุ้มครองให้มากขึ้นเพราะเว็บรู้ใจของเราจะทำการเปรียบเทียบราคาเบี้ยประกันในรูปแบบประกันที่คุณต้องการจากบริษัทประกันต่างๆและเลือกรูปแบบประกันที่มีราคาถูกที่สุดมาให้คุณได้รับความคุ้มครองความเป็นว่าถ้าใครสนใจต้องการเลือกซื้อบริการประกันใดๆก็ตามสามารถเข้ามาสอบถามรายละเอียดเพิ่มเติมกับเว็บรู้ใจของเราได้รวมถึงบริการเช็คเบี้ยประกันที่เว็บรู้ใช้ของเราก็พร้อมให้ความช่วยเหลือและให้คำแนะนำในการเลือกซื้อประกันสำหรับผู้ที่สนใจทุกท่าน

POCO F3 มือถือรุ่นใหม่ มาแรง ฟีเจอร์เด็ดระดับตำนานPOCO F3 มือถือรุ่นใหม่ มาแรง ฟีเจอร์เด็ดระดับตำนาน
บทความนี้เราจะมาพูดถึงตัวมือถือรุ่นใหม่ อย่าง POCO F3 กัน ซึ่งเราขอบอกเลยว่า รุ่นนี้เป็นรุ่นตำนานเลยก็ว่าได้ เพราะฟีเจอร์ที่เด็ดๆ ที่ทางแบรนด์งัดมาแต่ละตัวนั้นไม่ธรรมดาเลยจริงๆ ว่าแล้วก็อย่ามัวเสียเวลา เราไปดูกันเลยดีกว่าว่าฟีเจอร์ที่เด็ดๆ ที่ทางแบรนด์งัดมาแต่ละตัวนั้นมีอะไรที่น่าสนใจกันบ้าง POCO F3 มือถือรุ่นใหม่ มาแรง ฟีเจอร์เด็ดระดับตำนาน ฟีเจอร์เด็ดระดับตำนาน ของ POCO F3 คือ ชิปเซ็ตตัวแรง Snapdragon 870

เรื่องที่จะเกิดขึ้นเมื่อ “ดวงอาทิตย์” หายไปเรื่องที่จะเกิดขึ้นเมื่อ “ดวงอาทิตย์” หายไป
ดวงอาทิตย์นั้นเป็นหนึ่งในสิ่งสำคัญที่สุดในระบบสุริยะจักวาลเลยนะครับเพราะว่าให้ทั้งความร้อน ความสว่าง และ โลกของเรานั้นยังต้องพึ่งพาดวงอาทิตย์ด้วยเช่นกัน แต่ว่าทุกคนเคยสงสัยไหมครับ ว่าถ้าหากว่าดวงอาทิตย์ที่เรากำลังเห็นอยู่ทุกวันนั้น ถ้าหากว่าวันหนึ่งนั้นหายไป จะเกิดอะไรขึ้นบ้าง มาลองดูกันดีกว่านะครับว่าจะเกิดอะไรขึ้นบ้างถ้าหากว่าดวงอาทิตย์นั้นหายไป ไร้แดงดึงดูด อย่างที่เรารู้กันว่าโลกของเรานั้นหมุนรอบดวงอาทิตย์ แต่ว่าถ้าหากว่าโลกของเรานั้นไรดวงอาทิตย์นั้นจะทำให้โลกของเราไร้แรงดึงดูด หรือ ให้กล่าวคือ แรงดึงดูดของโลกนั้นหายไปเลยนะครับ และไม่ใช่แค่โลกของเรานะครับที่มีแรงดึงดูดจากดวงอาทิตย์ เพราะว่าดวงเคราะห์ดวงอื่น ๆ ก็ใชแรงดึงดูดจากพระอาทิตย์ที่เราเห็นอยู่ทุกวันด้วยเช่นกัน โลกจะมืดมิด อีกข้อนั้นคือในเรื่องของ “แสงสว่าง” ที่จะเกิดขึ้นอย่างแน่นอน ถ้าหากว่าพระอาทิตย์นั้นหายไป นั้นโลกของเรานั้นจะมืดลงนะครับ แต่ว่าไม่ใช่ว่าในแสงนั้นจะหายไปในทันทีนะครับ เพราะว่ายังมีการเดินทางของแสงของอยู่ แต่ว่า ในเวลาไม่นานนั้นแสงที่มีจะหมดไปอย่างแน่นอน และนั้นจะทำให้โลกทั้งใบของเรานั้นตกอยู่ในความมืดมิดทันที

ส่งสินค้าอย่างไรให้ลูกค้าประทับใจส่งสินค้าอย่างไรให้ลูกค้าประทับใจ
ปัจจุบันการขายสินค้าออนไลน์ถือเป็นสิ่งที่ได้รับความนิยมโดยทั่วไป เนื่องจากสมาร์ทโฟนเป็นอุปกรณ์อิเล็กทรอนิกส์ที่มีกันแทบทุกคนอยู่แล้ว เมื่อมีสมาร์ทโฟนอยู่ในมือ ทำให้หลายต่อหลายคนเลือกที่จะซื้อสินค้าออนไลน์แทนการไปจับจ่ายที่ห้างสรรพสินค้า เนื่องจากความสะดวกหลายประการ ทั้งรีวิวที่ตนเองนั้นสามารถดูได้อย่างง่ายดาย บวกกับการจ่ายสินค้าที่สะดวก รองรับการจ่ายเงินทุกรูปแบบ ไม่ว่าจะเป็นการจ่ายเงินปลายทาง การจ่ายผ่านบัตรเครดิต ในส่วนของพ่อค้าแม่ค้าออนไลน์เองก็ถือว่าต้องปรับตัวด้วยเช่นกัน เนื่องจากว่าปัจจุบันไม่ว่าใครต่างก็เป็นพ่อค้าแม่ค้าออนไลน์ได้ หากคุณเป็นอีกคนหนึ่งที่สงสัยว่าส่งสินค้าอย่างไรให้ลูกค้าประทับใจ ขอแนะนำดังนี้ 1.เลือกขนส่งที่จัดส่งเร็ว ปัจจุบันบริษัทขนส่งสินค้าออนไลน์มีหลายต่อหลายบริษัทให้ได้เลือกสรรกัน แต่ละบริษัทมีจุดเด่นแตกต่างกันไป บางบริษัทที่เปิดตัวใหม่ จะเน้นราคาที่ถูกเป็นหลัก แต่คุณภาพอาจไม่ได้ดีดังที่หลายคนปรารถนา หรือบางขนส่งจะเน้นการจัดส่งอย่างรวดเร็วและของไปถึงปลายทางโดยไม่บุบสลาย จึงไม่น่าแปลกใจเลยที่หลายคนจะเลือกการจัดส่งกับบริษัทที่มีคุณภาพ เพื่อให้ลูกค้าประทับใจนั่นเอง 2.การแพ็คสินค้า อีกหนึ่งสิ่งที่ลูกค้าต้องการในการซื้อสินค้าออนไลน์ก็คือการแพ็คสินค้า ส่วนใหญ่แล้วหากว่าเป็นเสื้อผ้า การแพ็คสินค้าไม่จำเป็นต้องมีกันกระแทกสักเท่าไรนัก บางคนอาจต้องการเพียงถุงซิปเท่านั้น ในขณะที่บางคนอยากให้ใส่เสื้อผ้าในกล่องกระดาษแทนการส่งในซองพลาสติกที่ใช้ได้เพียงแค่ครั้งเดียวก็ต้องทิ้ง ดังนั้นจึงเป็นสิ่งที่พ่อค้าแม่ค้าออนไลน์ต้องให้ความสำคัญและระมัดระวังอย่างมาก
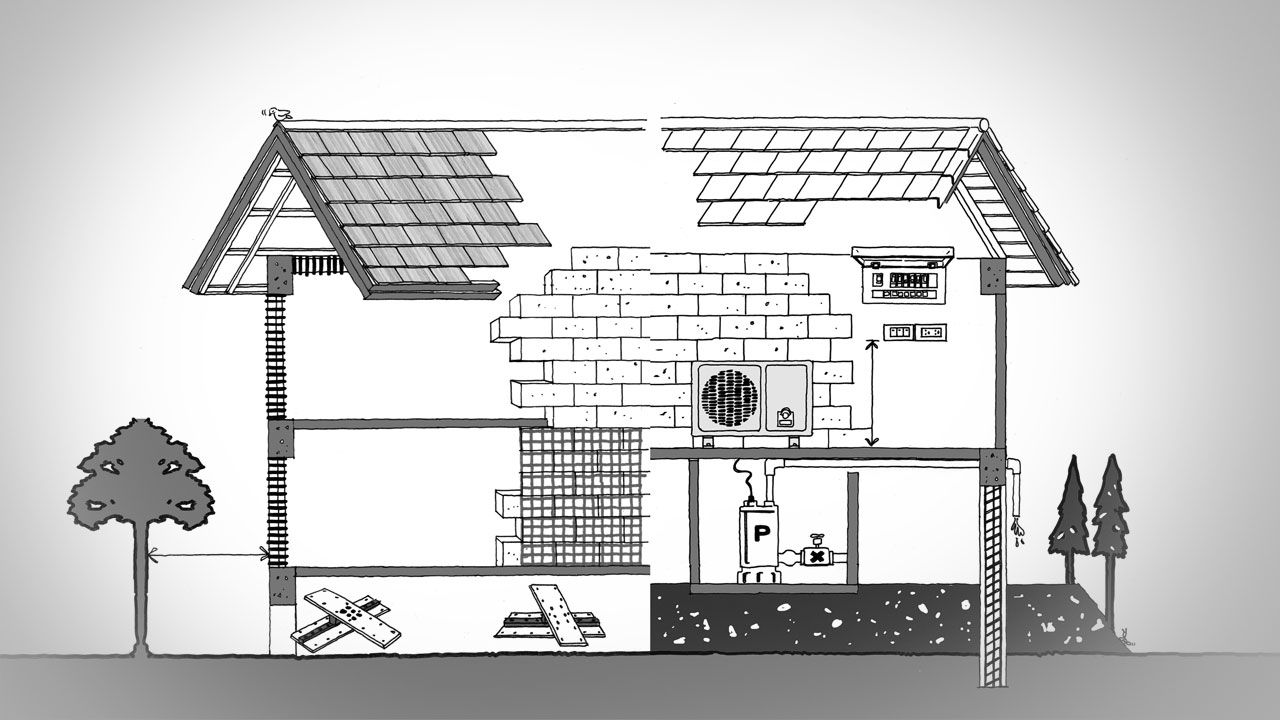
สร้างบ้านอย่างไร ให้ตัวบ้านแข็งแรงสามารถรับมือกับภัยธรรมชาติได้สร้างบ้านอย่างไร ให้ตัวบ้านแข็งแรงสามารถรับมือกับภัยธรรมชาติได้
สร้างบ้านอย่างไร ให้ตัวบ้านแข็งแรงสามารถรับมือกับภัยธรรมชาติได้ ทุกวันนี้ เราต่างต้องพบเจอกับภัยธรรมชาติมากมาย เพราะการกระทำของมนุษย์เอง ไม่ว่าจะเป็นการตัดไม้ทำลายป่า การเผาที่ สร้างโรงงาน เป็นต้น ฉะนั้น เราต้องเรียนตามตรงว่า เราทำอะไรมากไม่ได้ นอกจากวิธีการล้อมคอกเมื่อวัวหายนั่นเอง ซึ่งวิธีการล้อมคอกเมื่อวัวหายก็คือ เราต้องเรียนรู้วิธีการสร้างบ้านให้แข็งแรง ให้สามารถรับมือกับภัยธรรมชาติได้ โดยวิธีการสร้างบ้านให้แข็งแรง ให้สามารถรับมือกับภัยธรรมชาติได้ มีดังต่อไปนี้ สร้างบ้านอย่างไร ให้ตัวบ้านแข็งแรงสามารถรับมือกับภัยธรรมชาติได้ สร้างบ้านแข็งแรงให้รับมือกับภัยธรรมชาติด้วยการ : สำรวจพื้นดินที่ตั้ง ควรเลี่ยงปลูกบ้านบนพื้นที่ที่เป็นหินกรวด ทรายหรือดินเหลว ควรเลือกพื้นที่ที่เป็นชั้นหินแข็งแรง หรือชั้นดินที่หนาแน่น ลองสำรวจด้วยตาเปล่าก่อนขั้นแรก ถ้าจะให้แน่ใจควรให้วิศวกรเจาะสำรวจอีกครั้ง

เรื่องน่ารู้ก่อนไปหางาน HR managerเรื่องน่ารู้ก่อนไปหางาน HR manager
หากคุณเป็นอีกคนหนึ่งที่ชื่นชอบงานทางด้านฝ่ายบุคคล คงคิดใช่หรือไม่ว่าการทำงานทางด้านนี้ถือเป็นงานที่ท้าทายความสามารถอย่างยิ่ง เนื่องจากการทำงานที่เกี่ยวข้องกับคน ไม่ว่าในแง่ใด ล้วนแล้วแต่ต้องอาศัยทักษะในการเจรจาที่สูงอย่างยิ่ง สำหรับใครที่อยากหางาน HR manager แนะนำว่าควรศึกษาเกี่ยวกับตำแหน่งนี้ให้ถี่ถ้วนเสียก่อน และตำแหน่ง HR manager มีเรื่องที่น่ารู้ดังต่อไปนี้ 1.มีความสามารถในการพัฒนาคน เนื่องจากว่าตำแหน่งผู้จัดการเป็นตำแหน่งที่มีความรับผิดชอบสูง นอกจากจะรับผิดชอบลูกทีมในแผนกแล้ว ยังต้องรับผิดชอบการพัฒนาคนในองค์กรอีกด้วย เนื่องจากการพัฒนาศักยภาพของคนในองค์กรถือเป็นสิ่งจำเป็น ด้วยแนวคิดของฝ่ายบุคคลที่มองว่า “คน” คือหนึ่งในทรัพยากรที่มีคุณค่าไม่แพ้ทรัพยากรอื่นๆ 2.ประเมินผลงานของแต่ละคน นอกจากจะวางแผนเพื่อพัฒนาบุคคลในองค์กรแล้ว สิ่งที่ผู้จัดการฝ่ายบุคคลจะต้องทำก็คือการประเมินผลงานของแต่ละคน โดยส่วนใหญ่แล้วการประเมินผลงานมีเกณฑที่ตายตัวอยู่ แต่ต้องอาศัยดุลยพินิจในการตัดสินใจของผู้จัดการประกอบเข้าไปด้วยกัน ทั้งนี้ก็เพื่อเสริมสร้างและกระตุ้นแรงบันดาลใจในการทำงานให้มากยิ่งขึ้นกว่าเดิม ใครอยากหางาน HR manager ควรเตรียมประเมินผลงานเอาไว้จะดีที่สุด 3.มีความสามารถในการตัดสินใจ อีกสิ่งหนึ่งที่ถือว่าเป็นจุดสำคัญที่ต้องมีในผู้จัดการทุกคนก็คือความสามารถในการตัดสินใจ โดยอาจจะต้องตัดสินใจตั้งแต่เรื่องเล็กๆ ไปจนถึงเรื่องใหญ่ๆ เลยก็ว่าได้ บางเรื่องอาจมีผลต่อจิตใจ

วิธีการยืดอายุการใช้งานของมอเตอร์เกียร์ทดรอบวิธีการยืดอายุการใช้งานของมอเตอร์เกียร์ทดรอบ
มอเตอร์เกียร์ทดรอบ เป็นหหนึ่งในอปุกรณ์ของโรงงานอุตสาหกรรมที่ลาย ๆ คนนั้นอาจจะรู้จักกันดีอยู่แล้วใช่ไหมครับ? และเชื่อว่าหลายคนนั้นอาจจะยังไม่รู้ถึงวิธีการรักษา และ ยืดออายุการใช้งานของ มอเตอร์เกียร์ทดรอบ ดังนั้นในบทความนี้เราจะมาพูดถึง …วิธีการยืดอายุการใช้งานของมอเตอร์เกียร์ทดรอบ เพื่อที่เราจะสามารถใช้งานเกียร์ทดรอบได้นานขึ้นและช่วยประหยัดงบประมาณได้มากขึ้นด้วยนะครับ เลือกเกียร์ทดรอบให้ถูกต้องตามแบบที่เราใช้ อย่างที่รู้กันดีกว่า มอเตอร์เกียร์ทดรอบ นั้นจะมีการใช้งานที่หลากหลายแบบให้เราได้เลือกใช้งานกันดังนั้นก่อนที่เราจะเลือกซื้อมอเตอร์เกียร์ทดรอบนั้นเราจะต้องรู้ก่อนว่ามอเตอร์เกียร์ทดรอบ ของเรานั้นใช้การทำงานแบบไหน มีลักษณะแบบใด เพราะมอเตอร์เกียร์ทดรอบนั้นมีหลากหลายลักษณะ ไม่ว่าจะเป็นเฟืองตรง เฟืองเฉียง และอื่น ๆ อีกมากมาย นอกจากนั้นยังมีในเรื่องความเร็วอีกด้วยนะครับว่าเราจะต้อใช้ความเร็วแบบไหน ดังนั้นเราจะต้องรู้ก่อนว่าหน้างานของเรานั้นมีอายุการใช้งานแบบไหนนะครับ ห้ามโดนน้ำ แน่นอนว่าอุปกรณ์ไฟฟ้า และ มอเตอร์ไฟฟ้า หรือแม้แต่ มอเตอร์เกียร์ทดรอบ นั้นเป็นของที่ไม่ถูกกับน้ำอย่างยิ่งนะครับ ดังนั้นเราจะต้องรู้ไว้เลยว่าห้ามนำ มอเตอร์เกียร์ทดรอบ นั้นมาใช้งานกับน้ำ หรือโดนน้ำเด็ดขาดไม่อย่างนั้นอาจจะทำการทำงานของมอเตอร์เกียร์ทดรอบ นั้นรวนเอาได้ นอกจากนั้นยังมีในเรื่องของสนิมอีกด้วยที่ต้องระวัง ดังนั้นเราจะต้องระวังในเรื่องนี้ไว้ให้ดี ๆ นะครับ เพราะว่าถ้ากว่าโดนน้ำขึ้นมาอาจจะทำเสียายมากขึ้นได้นะครับ